


Unraveling the Complexity of Bioinformatics
The secret link between genomics, pathology, and drug discovery


TLDR: Bioinformatics has become an increasingly hot area in startup world. Automating and simplifying processes on the users’ side has become the goal of many companies that try to make the lives of bioinformaticians and their colleagues easier. Getting from raw lab data to insights has never been more complex, despite the flurry of available software tools — they are often academic and difficult-to-use legacy scripts. Beyond simplifying their use, the intersection between wet lab scientists and bioinformaticians poses its own unique challenges, which some startups are beginning to address. Despite the difficulties, companies with high expertise will have a lot of business to do in these spaces.
Authors’ note: If this newsletter is truncated in your email, you can click on "View entire message" and you’ll be able to view the entire post in your email app.
What do genomics, pathology, and drug discovery have in common? Apart from being performed by biologists and similar professions in R&D labs, they all rely on one thing: data. And this data is growing exponentially: Genomics data, for example, is doubling in size every seven months.
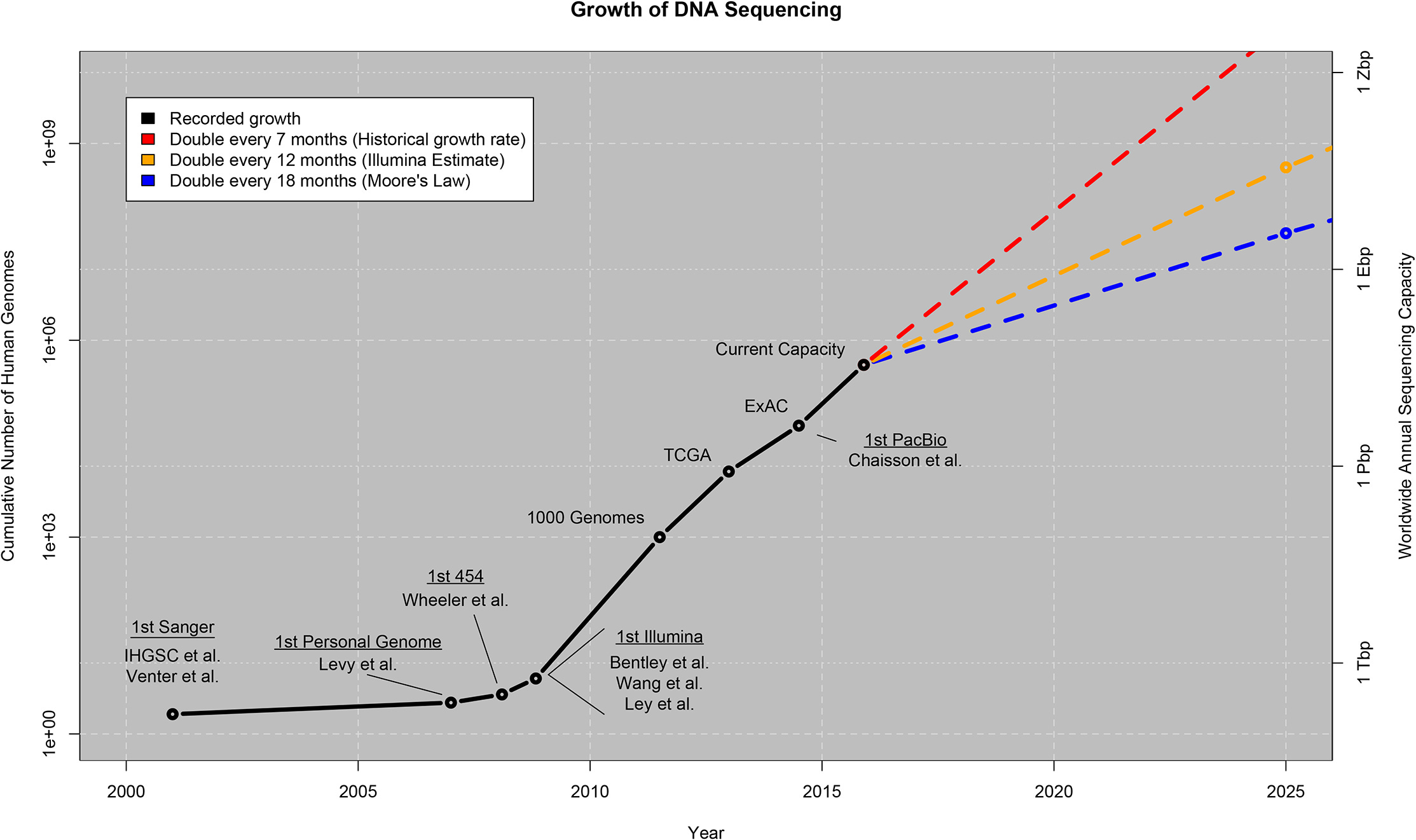
With this explosion of data, the collection, processing, storage, analysis, and publishing becomes more and more complex and requires many different mathematical and computational methods.
Many factors contribute to this complexity: Biological data, such as sequences, spectral data, images, or fluorescence intensity readings, is often voluminous and noisy. A full human genome, for example, can occupy 900 MB of disk space. Sophisticated algorithms are then required to extract insights from these often massive volumes of data.
Often, lab data also needs to be cleaned of any contaminations or other noise before further processing. The choice of algorithm, tool, or method, thereby depends on the data at hand and the research questions. Finally, many experiments are repeated across different labs, but biological databases are often not equipped for interoperability. This means that a lot of information stays inside silos, which limits research productivity and output.
What is a bioinformatician?
A bioinformatician is a scientist who applies computational techniques and methods to analyze and interpret biological data, particularly in the fields of genetics, genomics, and molecular biology. They use their expertise in biology, statistics, computer science, and mathematics to develop and/or use algorithms, software tools, and databases to extract meaningful insights from large sets of biological data, such as DNA sequences, protein structures, and gene expression profiles.
Bioinformaticians collaborate closely with wet lab scientists (experimental biologists who work directly with biological materials in laboratory settings). Wet lab scientists typically generate data in the lab that gets processed and analyzed by bioinformaticians in an intertwined process with many feedback loops.
From lab data generation to interpretation
More often than not, getting biological data from a wet lab to a publishable analysis is a lengthy and complex process.
Data generation
Researchers typically prepare samples of tissue or other biological material, and then analyze them using various techniques and lab machines, depending on the type of data that they need. Such techniques include:
Sequencing of a whole genome, exome, or RNA for genomic and transcriptomic data,
Mass spectrometry for proteomics (i.e., the study of the interactions, function, composition, and structures of proteins and their cellular activities) and for metabolomics data (i.e., the measurement of all metabolites and low-molecular-weight molecules in a biological specimen),
Microscopy for cellular or subcellular imaging, and
Flow cytometry for cell sorting and counting specific cell types based on their properties.
Each of these techniques generates raw data in the form of sequences, spectral data, images, fluorescence intensity readings, and other formats.
Data pre-processing
Raw data often requires pre-processing to remove noise and artifacts, i.e., any distortions or anomalies in the data that do not represent the biological signal or phenomenon being studied. Such artifacts can originate from the specifics of the equipment used, or from contamination, among other sources.
Contaminated or low-quality data must therefore be filtered out, by hand or by using dedicated algorithms and tools. Sequences need to be aligned to reference genomes to correct for any sequencing errors. Statistical methods are used to adjust for background noise and to normalize the data. Depending on the type of data and its quality, other steps might also be part of the process.
Data storage
Both the raw and pre-processed data is stored in databases which are designed to handle large volumes of biological data. Storing data often involves annotating the data with metadata that describe the experimental conditions, sample origins, and analysis parameters.
Data analysis
The data then gets analyzed with the use of bioinformatics tools and statistical methods. The choice of such tools and methods depends on the type of data at hand and on the research questions. Some examples include identifying differentially expressed genes, detecting genetic variants, quantifying protein abundances, or characterizing cellular phenotypes.
Publication and data sharing
Finally, the results are compiled, visualized, and interpreted in the context of the original research questions. Scientists can publish their findings in scientific journals, often adding the raw and processed data in public databases to enable further research by the scientific community. This last step is optional for companies which have a profit motive and hence have incentives to keep their findings hidden from the competition.
An introduction to bioinformatics pipelines
Bio pipelines are automated workflows that systematically process large datasets through various analysis tools and software. They take over after the wet lab experiments are completed, and aim to make data pre-processing, analysis, and interpretation more efficient.
Aside from making research less labor-intensive, this has another interesting side effect: Pipelines standardize the processing and analysis steps, making the research more reproducible and transparent. This greatly helps ensure that the results can be validated by other scientists, and makes data across different studies more comparable.
With user permission, such pipelines can also aggregate data from different sources and experiments, and integrate them into the final analysis. This might involve combining genomic, transcriptomic, and proteomic data to understand a disease mechanism or cellular responses to treatments. Aggregation hence allows a more comprehensive analysis, and can be prohibitively lengthy without data pipelines.
Automation and standardization, tailored to each type of data
A wide array of techniques exist to automate biological data treatment. These are not all full pipelines: Many of them only automate a part of the process, and can be put together to assemble a full pipeline. Below is a list of popular tools, which is by no means complete — many more tools exist for specific subdomains.
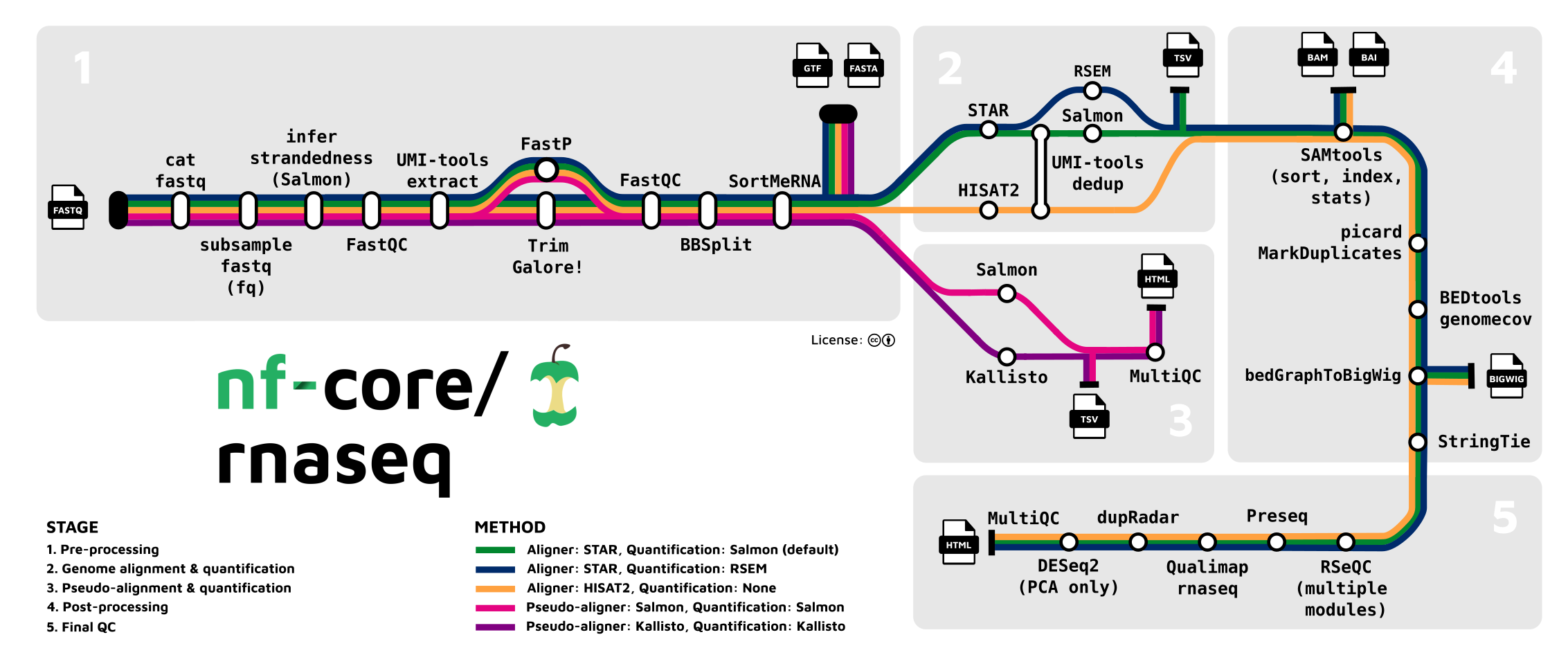
nf-core/rnaseq: This is a bioinformatics pipeline that can be used to analyse RNA sequencing data obtained from organisms with a reference genome and annotation. It takes a samplesheet and FASTQ files as input, performs quality control (QC), trimming and (pseudo-)alignment, and produces a gene expression matrix and extensive QC report.
Salmon: It uses quasi-mapping with a two-phase inference procedure to quantify the expression of transcripts using RNA-sequencing data. Salmon uses an expressive and realistic model of RNA-sequencing data that takes into account commonly observed experimental attributes and biases.
RoseTTAFoldNA: This is a deep-learning approach to modeling of nucleic acid and protein-nucleic acid complexes. It rapidly produces 3D structure models with confidence estimates for protein-DNA and protein-RNA complexes, and for RNA tertiary structures.
Trim Galore!: It is a wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files, with some extra functionality for MspI-digested RRBS-type (Reduced Representation Bisufite-Seq) libraries.
MethylKit: This R package rapidly analyzes genome-wide cytosine epigenetic profiles from high-throughput methylation andhydroxymethylation sequencing experiments. DNA methylation is is a chemical modification of cytosine bases that is pivotal for gene regulation, cellular specification and cancer development.
rnafusion: This tool is used on RNA-Seq data with the intention of localising and visualising fusion genes. The pipeline uses Nextflow, a bioinformatics workflow tool. It pre-processes raw data from FastQ inputs, aligns the reads and performs extensive quality-control on the results. The workflow is built on a list of curated tools such as Arriba, EricScript, and FusionCatcher.
RSEM: This is a software package for quantifying gene and isoform abundances from single-end or paired-end RNA-Seq data. It is a very popular and well-cited tool.
QIIME 2: This platform for microbiome bioinformatics is extensible, free, open source, and community developed, according to its authors. One of the more complete tools, it enables researchers to start an analysis with raw DNA sequence data and finish with publication-quality figures and statistical results.
BLAST: This Basic Local Alignment Search Tool finds regions of similarity between biological sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance.
BWA: The Burrows-Wheeler Alignment tool aligns short sequencing reads against a large reference sequence such as the human genome, allowing mismatches and gaps. This is extremely useful for short reads of DNA sequencing, which allows to check the genome for specific characteristics, for example hereditary diseases.
**CRISPresso2:** This pipeline is built for the analysis of genome editing experiments. It is designed to enable rapid and intuitive interpretation of results produced by amplicon sequencing.
STAR: The Spliced Transcripts Alignment to a Reference (STAR) tool is designed to align short reads to a reference genome. It is an open-source C++ code that uses sequential maximum mappable seed search in uncompressed suffix arrays followed by seed clustering and stitching procedure.
PIPseeker: One of the few commercial codes on this list, this tool helps analyze sequencing data from PIPseq Single Cell Gene Expression libraries. The software makes it easy to obtain summary metrics, diagnostic plots, clustering and differential gene expression tables.
Ampliseq: This is a bioinformatics analysis pipeline used for amplicon sequencing (an amplicon is a piece of DNA or RNA that is the source or product of amplification or replication events). The tool supports denoising of any amplicon, and can work with a variety of databases for taxonomic assignment.
MultiQC: This tool a creates a single report visualising output from multiple tools across many samples, enabling global trends and biases to be quickly identified. It can plot data from many common bioinformatics tools and is built to allow easy extension and customization.
alevin-fry: This tool can be used to quantify sc/snRNA-seq data, and also how the spliced and unspliced molecule quantification required as input for RNA velocity analyses can be seamlessly extracted from the same preprocessed data used to generate normal gene expression count matrices.
DADA2: This is an algorithm for accurate high-resolution of sample composition from amplicon sequencing data. It is very well established within the bioinformatics community, having received over 14,000 citations to date.
BedTools: This is a suite of tools aiming at testing for correlations between different sets of genomic features, and ultimately being able to compare different genomic features. BedTools is also very well established with over 20,000 citations to date.
viralrecon: This is a bioinformatics analysis pipeline used to perform assembly and intrahost or low-frequency variant calling for viral samples, notably SARS-Cov2. The pipeline supports metagenomics and amplicon sequencing data derived from the Illumina sequencing platform.
SAMtools: The SAM format is a generic alignment format for storing read alignments against reference sequences. SAMtools implements various utilities for post-processing alignments in the SAM format, such as indexing, variant caller and alignment viewer.
DeepVariant: This is an open-source cohort-calling method, which enables analysis of population-scale sequenced cohorts in order to identify variants informative for diseases, traits and ancestry. This is important given the volume of data that must be processed on a cohort level.
Bowtie 2: This tool tackles aligning sequencing reads to a reference genome. It boasts an effective combination of speed, sensitivity and accuracy across a range of read lengths and sequencing technologies.
GATK: The Genome Analysis Toolkit is a structured programming framework designed to enable the rapid development of efficient and robust analysis tools for next-generation DNA sequencers. Its subtools were built with a primary focus on variant discovery and genotyping in mind.
LeafCutter: This is an algorithm for outlier splicing detection in rare diseases. It is geared towards identifying RNA splicing outliers, which is particularly useful for determining causal Mendelian disease genes.
Dorado: This is a data processing toolkit that contains Oxford Nanopore Technologies' basecalling algorithms, and several bioinformatic post-processing features. Such basecalling algorithms learn how to determine nucleotide sequence via machine learning.
cutadapt: This is a tool for quality control of high-throughput sequencing reads. The functions include an adapter, primer, and poly-A tail removal.
vcftools: VCF is a generic format for storing DNA polymorphism data such as SNPs, insertions, deletions and structural variants, together with rich annotations. VCFtools is a software suite that implements various utilities for processing VCF files, including validation, merging, comparing and also provides a general Perl API.
As you might see, there are many different tasks, which are dealt with by different tools. Putting all these tools together to create some standard go-to solution is far from evident.
Challenges at the intersection between wet lab scientists and bioinformaticians
Data storage and provenance is one of the key challenges, because many research teams currently store lab raw data and processed data formats in simple folder systems. This often happens without metadata management or audit trails on the processing steps that transformed the raw data. This results in situations in which it is very hard to find the right data for an analysis task or reproduce experiments.
As mentioned earlier, the pipelines themselves are often complex and difficult to use. For researchers without much coding expertise, this can be especially challenging: The tools are often developed by academics, decades old, not user-friendly, and require significant training to use effectively. They can therefore not be run by wet lab scientists, which is pretty much the raison d’être of bioinformaticians.
Biological analyses are naturally voluminous, complex, and large-scale. This means that computational requirements are often high. Managing these requirements and efficiently processing data is a challenge for many research teams. Although many pipelines are designed to work quicker through large datasets and minimize computing requirements, it remains a big problem in the field. There is also a high variability of computational workloads: computing capacity needs tend to have large spikes that are interspersed quite irregularly over time. Labs or downstream data analytics providers typically run an experiment (heavy load) and then don’t run compute-intense stuff for a couple of weeks until the next experiment or pipeline needs to be run.
Because both biological lab analyses and the processing of biological data are so complex, there is a knowledge gap between wet lab researchers and bioinformaticians. This gap creates friction and inefficiencies, slowing down the research process and complicating data management and analysis. Additionally, bioinformaticians are a pretty scarce resource, which adds another bottleneck to an already strained field.
All these factors make the field challenging, but also exciting. If one thing is clear, it is that more and better bioinformatics pipelines are still needed in order to accelerate research in a rapidly aging society. The future thus remains equally complex and exciting.
Interesting startups in the bio data space
As the intersection of technology and biology continues to shape the future of the life sciences and drug development industries, it is super interesting to understand the evolving startup market landscape.
The venture fund MMC made a great comprehensive overview of this landscape in a report by Charlotte Barttelot. Mostly relevant for our analysis are the subsections about ‘lab operations and data management’ as well as ‘data infrastructure and analysis’:
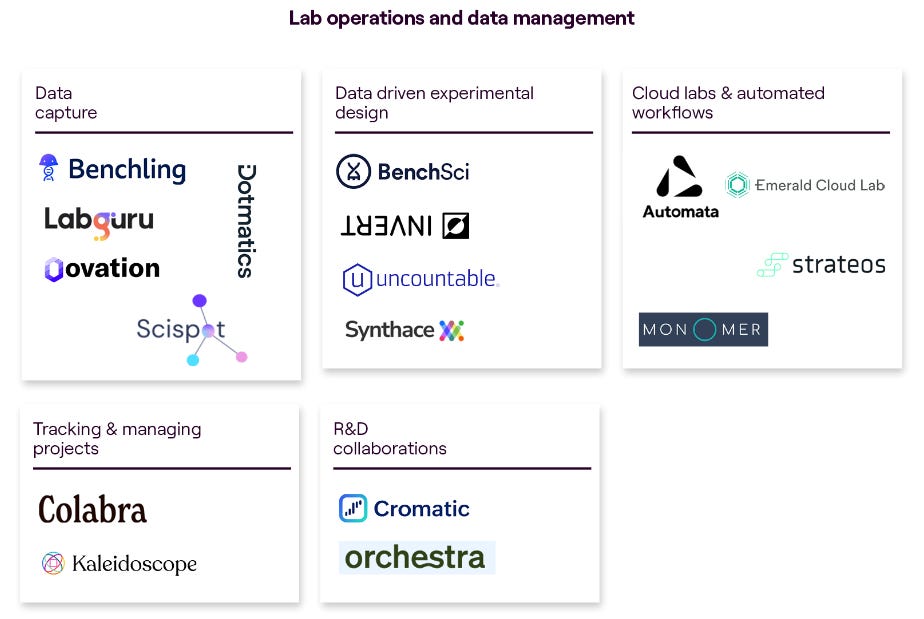
Some particularly interesting companies that we looked at in detail during our research:
Benchling is a software company that specializes in providing cloud-based solutions for life sciences research and development. Their platform offers tools for data management, experimental design, workflow automation, and collaboration, tailored specifically for biotech, pharmaceutical, and academic research organizations. They started their journey with an electronic lab notebook and then expanded into other verticals.
They have raised over 400m$ over the last years from top investors like Andreessen Horowitz, Benchmark, Altimeter and Sequoia.
Nextflow is not actually a commercial company. Nextflow is an open-source framework designed to facilitate the development and execution of computational pipelines. Nextflow simplifies the creation of reproducible and scalable workflows by providing a domain-specific language (DSL) that allows researchers to define their analysis pipelines using familiar syntax. It offers features such as parallel execution, fault tolerance, and support for various computing environments, including local machines, clusters, and cloud platforms.
Nextflow is the de-facto standard on how to build pipelines in the bioinformatics world and is used by tens of thousands bioinformaticians, clinicians, and data scientists worldwide.
The founders of Seqera Labs started the Nextflow project back in 2013 during their studies at Barcelona's Centre for Genomic Regulation (CRG) and started Seqera as a commercial endeavor on top of Nextflow.
They essentially offer a managed version of Nextflow and other tools in the bioinformatics space to make it easier for companies to manage the computational complexities behind running large scale pipelines. They are also increasingly expanding into adjacent data exploration workflows.
They have raised over ~30m$ over the last years from top investors like Speedinvest, AminoCollective and Addition.
Some emerging players that were not part of the report by MMC are e.g. flow.bio and mantle.bio (part of the current YC batch).
More exciting tech news
Beautifully put together,
’s 10 Charts That Capture How the World Is Changing serves insights on a silver palette. To name just a few: AI is already surpassing humans in almost every discipline, electric vehicles are booming, and AI is becoming a huge boon to the gaming industry.Tips for LLM Pretraining and Evaluating Reward Models is a clear and succinct roundup on the state of the art of handling LLMs smartly.
treats a complicated topic in a compelling way and makes it understandable for the average engineer. We also thoroughly enjoyed his list of interesting new papers.The hardware enthusiasts among you might enjoy this piece: Why Did Supersonic Airliners Fail? by
explains just how risky deviating from the status quo in aviation really is, and how pushing technology with increasing amounts of capital has its limits. The proof? We haven’t had another supersonic aircraft for the past fifty years, and this is not for lack of trying.